RiboSketch
Bond editing, E. Coli 5S rRNA with SHAPE data coloring
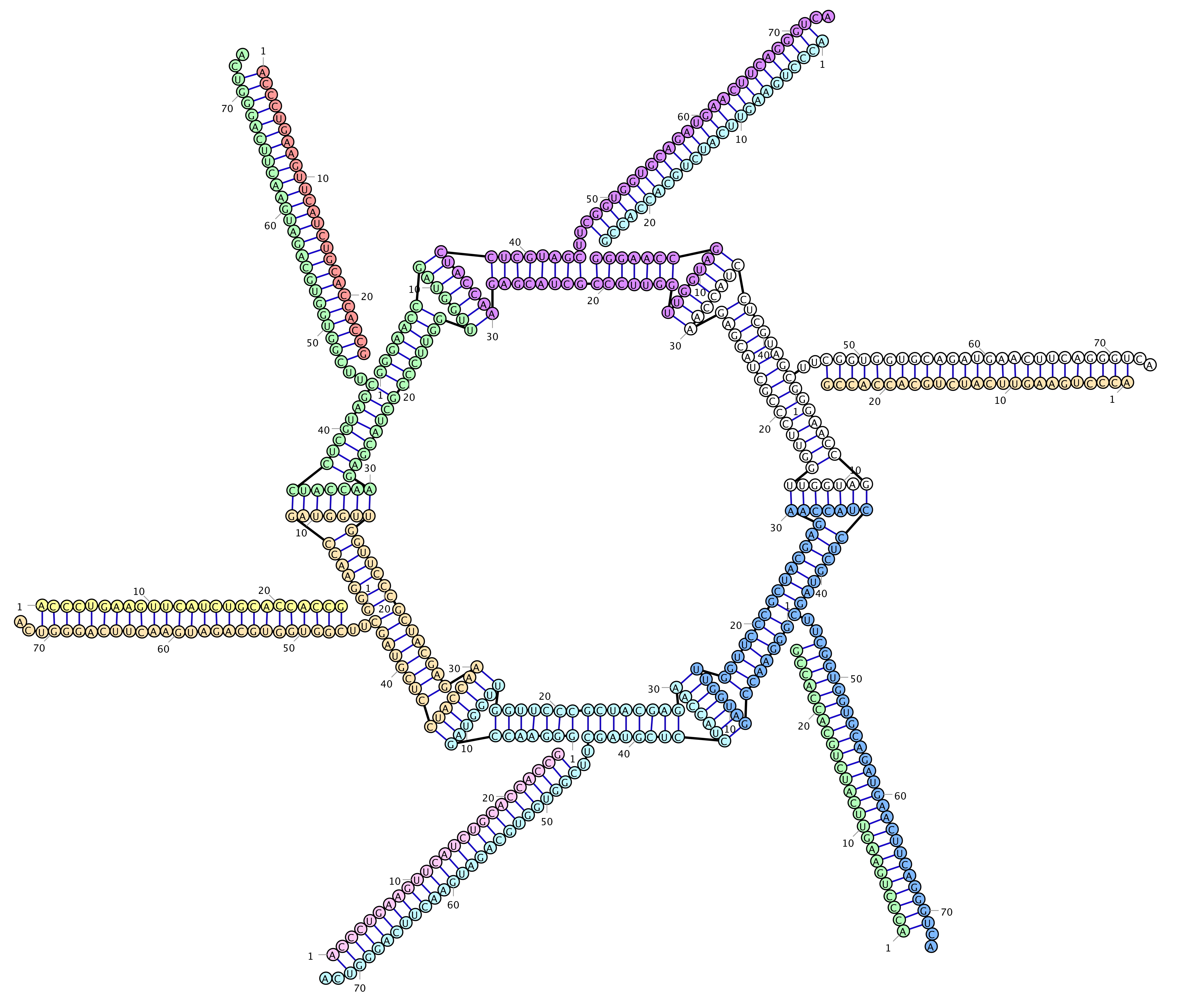
Hexameric Ring with arms
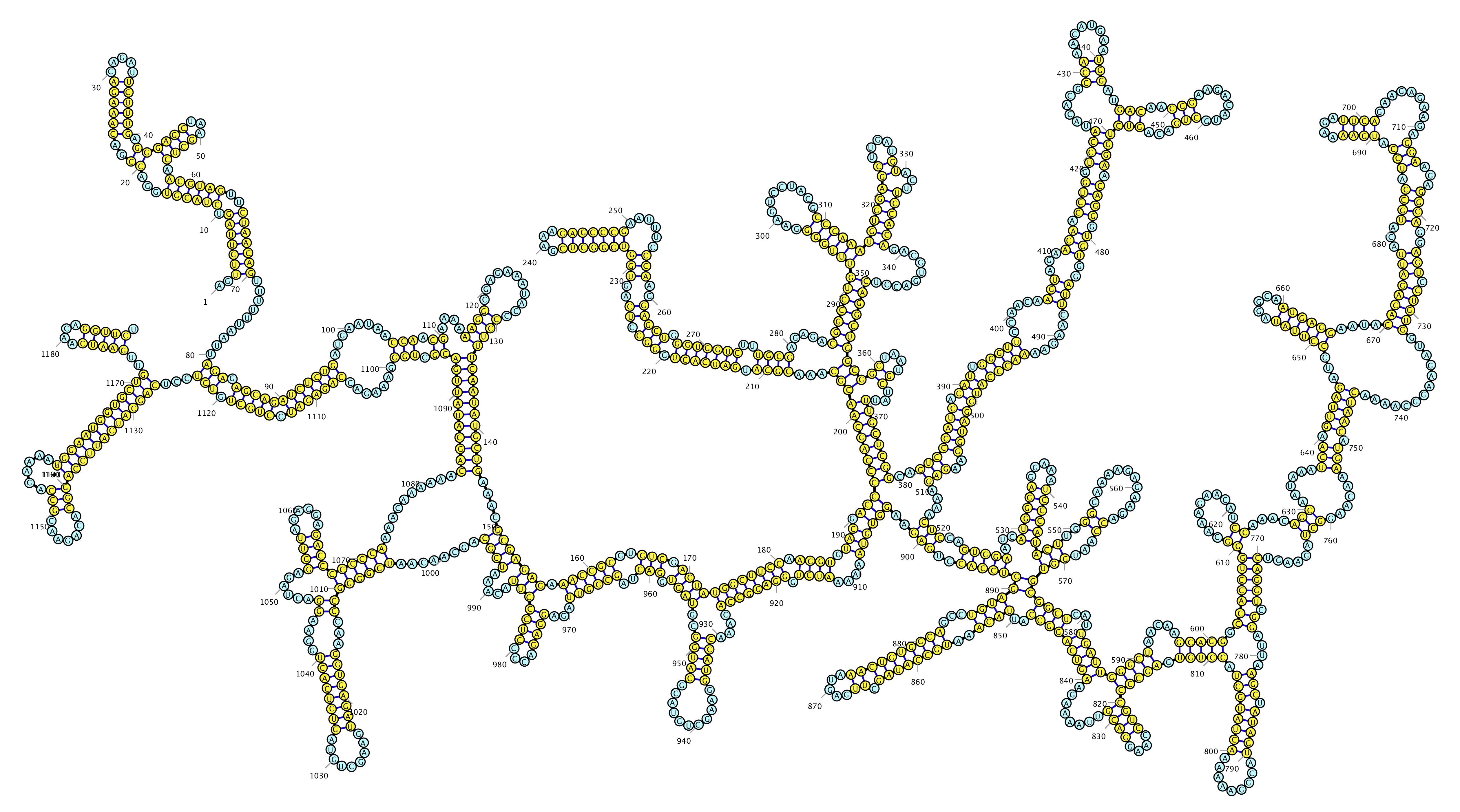
Dengue Virus

Hexameric Ring, dynamic formation
Bioinformatics Publication
If you use RiboSketch in your scientific work, we would greatly appreciate a citation to our publication.
What is RiboSketch?
- RiboSketch is a drawing program for the production of RNA and DNA secondary structure images.
- The user provides an input file (.ct, .bpseq, .dbn, or the save file type .rs) containing the sequence and base-pairing of the strand(s).
Features
- Works with multiple strands, non-canonical interactions, hybrid RNA-DNA base-pairing, pseudoknots, and interstrand interactions
- Creates automatic layouts and circle diagrams
- Dynamic simulation mode
- Interactive editing is enabled through precise commands
- The user may save the state of the program and load previous save-states
- Actions may be undone and redone
Using the Program
- Load a secondary structure file (you can add additional bonds and non-canonicals once in the program)
- Default start state is the “Radial Layout”.
- Mouse over the top of the screen to access the MENU and to view COMMANDS.
Mouse Commands
- Click on (or drag a selection box over) bases to select them. Hold shift to multi-select or toggle the selection of bases.
- Drag selected nucleotides to move them.
- To select individual nucleotides, hold ALT/OPTION and click.
- To select one half of a helix, you can either:
- Select the helix, then shift click on the side you don't want to unselect it.
- Click and drag a selection box over one side of the helix.
- ROTATION: “m” rotates clockwise about the mouse cursor, “n” counterclockwise. Hold shift to rotate with a smaller angle.
- Select the helix, then shift click on the side you don't want to unselect it.
- Click and drag a selection box over one side of the helix.
Menu Features
Sliders
- Base Size: Size of nucleotides
- Bond Length: Distance between base-paired nucleotides
- Chain Length: Effective only during simulation mode, controls distance between adjacent nucleotides
- Color Scheme: “Pastel”, “White”, "Light", “Bright”, “Grey", and special ones:
- Base Type: Each nucleotide type gets its own color
- Rainbow: Each individual base gets its own color
- Structure: Paired bases: Yellow, Unpaired: Blue
- Custom: User must input number values corresponding to each base
- Base Type: Each nucleotide type gets its own color
- Rainbow: Each individual base gets its own color
- Structure: Paired bases: Yellow, Unpaired: Blue
- Custom: User must input number values corresponding to each base
Buttons
- Save: Writes state of program into a text file (.rs), which can be loaded into RiboSketch.
- Load File: Read a new secondary structure file into the program.
- Load Bonds: Add new bonds from a text file of comma-separated entries with format: “firstID secondID bondType”
- Example: 3 20 cWW, 4 19 tHS, 6 30 cSS
- Load Colors: Color nucleotides based on a space-separated list of numbers (probing data) from a text file.
- Radial Layout: Position nucleotides with the NAView algorithm, expanded to accommodate multiple strands and pseudoknots.
- Circle Layout: Nucleotides are positioned in a circle with base pairs drawn as chords.
- Simulation Mode: Apply forces.
- Sim. Selected Mode: Only apply forces to selected nucleotides.
- Rigid Helices: Enforce right angles for helices.
- Rigid Loops: Circularize loops and straighten stems.
- Rigid Hairpins: Circularize hairpin loops. Only has effect when Rigid Loops is OFF.
- Zoom Reset: Return zoom to default.
- Outlines: Toggle drawing circles around nucleotides.
- Labels: Display numbering for every tenth nucleotide.
- PNG Screenshot: Saves a screen image to the folder of the input file.
- SVG Screenshot: Saves an SVG file of the screen to the folder of the input file.
- Example: 3 20 cWW, 4 19 tHS, 6 30 cSS
Keyboard Commands
Editing
- ROTATION: “m” rotates clockwise about the mouse cursor; “n” counterclockwise. Hold shift to rotate with a smaller angle.
- "z": UNDO. Shift-Z to REDO.
- "s": Turn on or off SIMULATION (forces)
- "S": Toggle the SIMULATION MODE to apply forces to selected nucleotides only or all nucleotides.
- “f”: FLIP the halves of a selected helix.
- “x” or “y”: Flips the selected bases over the X or Y-axis.
- "r": RELAX FORCES for selected nucleotides. Effective only in Simulation mode.
Display
- + / - : Zooms the screen in or out
- 0 : Resets the view scale
- Arrows : When no nucletodies are selected, the arrow keys translate the window view.
- 1 / 2 : Brings the selected bases to the front or back, akin to Powerpoint
- b / B : Base SIZE +/- (Same as slider)
- o : Toggle OUTLINE
- i : Toggle LETTERS
- l : Toggle LABELS (Same as button)
- k : View base number in secondary file
- v : Toggle ANNOTATIONS (5' and 3', strand number)
- c : Changes the color scheme of the nucleotides
- p : Saves a PNG SCREENSHOT (Same as the button)
Adding and Deleting Bonds
- Adding: Hold Spacebar, click first base, click second base.
- Non-Canonical: Click "Load Bonds" button to add bonds of a specified type through text input (Ex: 3 25 cWW, 8 12 tHS)
- Deleting: Hold "d", click a base to delete its bonds.
The "RS" Data Format
- sequences: semicolon-separated list of RNA or DNA sequence strings which are in upper or lower case respectively
- starts: comma-separated list of zero-based indices of the first residue of each sequence
- positions: semicolon-separated string of x,y,z positions of each residue. In the current version the z-values are set to zero.
- radius: the radius of the residues in pixel units
- backbone_distance: target distance of two residues that are adjacent in sequence
- basepair_distance: target distance of two base paired residues
- color_scheme: an integer that stands for the color mode
- outline: true/false indicating whether the residues are drawn with a black circle
- movement: true/false indicating whether the simulation mode is on
- labels: true/false indicating whether the residue labels are shown or not
- rigid_helices: true/false indicating whether rigidifying helices is activated or not
- rigid_loops: true/false indicating whether rigidifying loops is activated or not
- rigid_hairpins: true/false indicating whether rigidifying hairpins is activated or not
- relax_ids: comma-separated list of residues whose basepairs are not affected by forces
- display_order: residues are displayed in this order. This is useful in order to achieve foreground/background effects
- pair_table: residue partners for each base pair (-1 for unpaired bases). Similar to base pair column in a CT format file.
- non_canonicals: extra information about non-canonical base pairs
- base_colors: assignable colors for each base (comma-separated RGB values between 0 and 1)