StructureLab is a computational system which has been developed to permit the use of a broad array of approaches to the analysis of the structure of RNA. The goal of the development is to provide a large set of tools that can be well integrated with experimental biology to aid in the process of the determination of the underlying structure of RNA sequences (Shapiro and Kasprzak 1995, Shapiro and Kasprzak 1996, Kasprzak and Shapiro 1999, Shapiro, Kasprzak, Grunewald, Aman 2006).
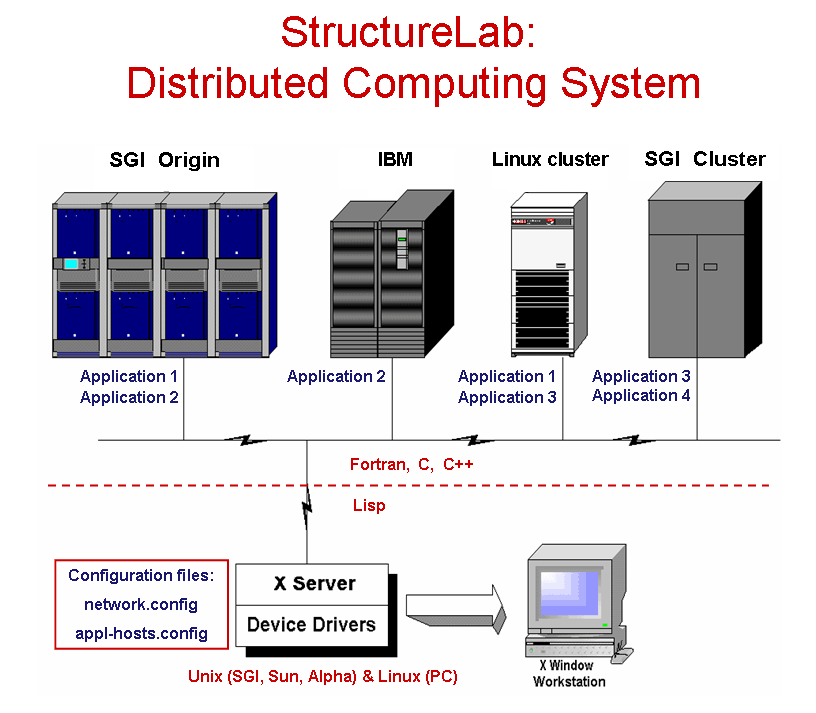
The approach taken views the structure determination problem as one of dealing with a data base of many computationally generated structures and provides the capability to analyze this data set from different perspectives. Many algorithms are integrated into one system which also utilizes a heterogeneous computing approach permitting the use of several computer architectures to help solve the posed problems. These different computational platforms make it relatively easy to incorporate currently existing programs as well as newly developed algorithms and to match these algorithms to the appropriate hardware. The system has been written in Common Lisp running on SGI, SUN, and Alpha Unix workstations, as well as PCs running Linux. It may also use a PC as a display devices, if it has an appropriate X-windows emulator software.
StructureLab utilizes a network of participating machines defined in reconfigureable tables. A window based interface makes this heterogeneous environment nearly transparent to the user.
The figure (above right) is a general view of some of the available workbench tools in StructureLab (clockwise); the main menu, the taxonomy tree windows, the 2D stem histogram, the Stem Trace control window and a multple solution space plot (data from our massively parallel genetic algorithm, MPGAfold), the structure drawing with base labeling, amino acid labeling, and annotations, a small color scale window, and the large-scale structure drawing.
The following list of specific functional domains is meant to illustrate the major capabilities of the system. It is not an exhaustive list of functions available in STRUCTURELAB.
Several real-time applications running on the user's workstation accept, manipulate, and return nucleic acid sequence strings (strings of characters). Functions available in this group perform sequence creation, manipulation via several types of mutations (single or many bases), and translation to amino acids sequences . A typical sequence format, among several possible, is illustrated below:
;test.seq file - sequence in STANFORD format
m2
GAAUUACCGAUAUCGAUACAUCAGGAAUAUUUGAUUCAGAUGAUAUGACUAUCAAGGCCG
CCUGAGUGCGGUUUUACCGCAUACCAAUAACGCUUCACUCGAGGCGUUUUUCGUUAUGUA
UAAAUAAGAAGCACACCAUGCAAUAUGCCAUUGCAGGGUGGCCUGUUGCUGGCUGCCCUU
CCGAAUCUUUACUUGAACGAA1
A CT file format (MFOLD 3.0+ output format) that contains both sequence and structure information, is illustrated below:
If the value of 5', 3' or paired base index is zero, it means that the nucleotide is not connected or paired with anything.
314 dG = -98.3 rabbit-RBG-mRNA
| 1 | G | 0 | 2 | 47 | 10 |
| 2 | G | 1 | 3 | 46 | 11 |
| 3 | G | 2 | 4 | 45 | 12 |
| 4 | A | 3 | 5 | 0 | 13 |
| 5 | G | 4 | 6 | 43 | 14 |
| : | : | : | : | : | : |
Folding programs employ two different types of algorithms; the Dynamic Programming Algorithm (DPA) (references)and the Genetic Algorithm (GA)(references). While both attempt to predict secondary structures of an RNA sequence, they differ in basic concepts used. The RNA folding algorithms accept sequence files (strings) as input, and output multiple region tables indicating which bases (nucleotides) are paired in a folded structure. These region tables reflect energetically optimal and suboptimal solutions based on standardized energy rules.
| Region Number | Start (First 5' base) | Stop (Last 3' base) | Region size (# of base pairs) | Energy (kcal/mol) |
|---|---|---|---|---|
| 1 | 9 | 53 | 4 | -4.3 |
| 2 | 13 | 48 | 2 | -2.3 |
| 3 | 16 | 46 | 3 | -2.0 |
| : | : | : | : | : |
Structural representation for a set of RNA molecules can be created based on the region files created by folding programs. Secondary (2D) structures can also be represented as trees, utilizing Lisp's nested list notation, with symbols such as M (multibranch loop), B (bulge loop), I (internal loop), and H (hairpin loop). Optional R's present in some representations are no-op place holders indicating regions (hence R) or stems. The tree representation facilitates multiple levels of abstraction of the actual structure allowing for structural comparisons of varying strictness by taxonomy tree clustering, based upon measures of structure similarity. In addition, multiple alignment methods and structural motifs matching can be employed.
Two of the Tree List representations used are shown below:
(N(H)(H)(BH)(H)(H)(H)(BBBIH)) - condensed
(N(R(H))(R(H))(R(B(R(H))))(R(H))(R(H))(R(H)) - expanded (explicit)
(R(B(R(B(R(B(R(I(R(H)))))))))))
{kind=link}