Stem Trace: The Concept
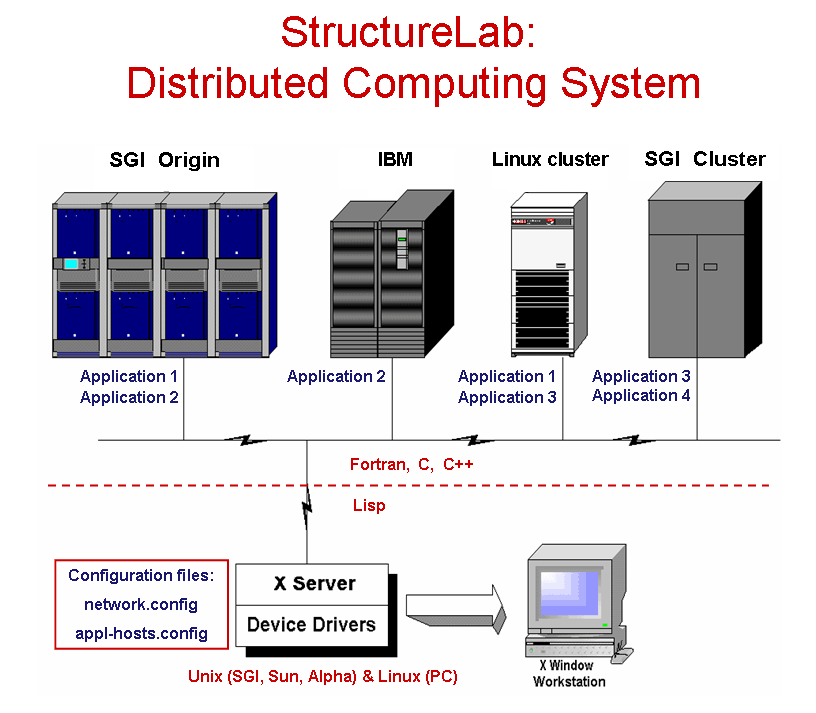
Structure Lab ToolsStructureLab is a computational system which has been developed to permit the use of a broad array of approaches to the analysis of the structure of RNA. The goal of the development is to provide a large set of tools that can be well integrated with experimental biology to aid in the process of the determination of the underlying structure of RNA sequences (Shapiro and Kasprzak 1995, Shapiro and Kasprzak 1996, Kasprzak and Shapiro 1999, Shapiro, Kasprzak, Grunewald, Aman 2006).
The approach taken views the structure determination problem as one of dealing with a data base of many computationally generated structures and provides the capability to analyze this data set from different perspectives. Many algorithms are integrated into one system which also utilizes a heterogeneous computing approach permitting the use of several computer architectures to help solve the posed problems. These different computational platforms make it relatively easy to incorporate currently existing programs as well as newly developed algorithms and to match these algorithms to the appropriate hardware. The system has been written in Common Lisp running on SGI, SUN, and Alpha Unix workstations, as well as PCs running Linux. It may also use a PC as a display devices, if it has an appropriate X-windows emulator software.
{kind=link}
StructureLab utilizes a network of participating machines defined in reconfigureable tables. A window based interface makes this heterogeneous environment nearly transparent to the user.
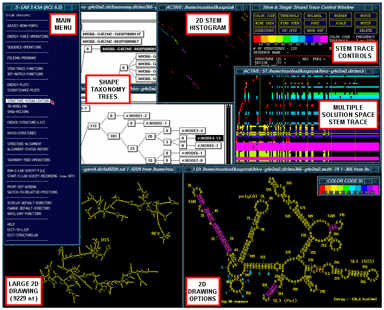
The figure (above right) is a general view of some of the available workbench tools in StructureLab (clockwise); the main menu, the taxonomy tree windows, the 2D stem histogram, the Stem Trace control window and a multple solution space plot (data from our massively parallel genetic algorithm, MPGAfold), the structure drawing with base labeling, amino acid labeling, and annotations, a small color scale window, and the large-scale structure drawing.
Specific Capabilities
The following list of specific functional domains is meant to illustrate the major capabilities of the system. It is not an exhaustive list of functions available in STRUCTURELAB.
- Sequence Manipulation
- Folding Algorithms
- Structure Representations
- Structure Alignment
- Taxonomy Operations
- Structure Matching
- RNA Structure Drawing and Manipulation (2D and 3D)
- Two-Dimensional Stem Histogram
- Stem Trace
- Tertiary Interactions Prediction
- Significance/Stability Plots and Energy Plots
- Miscellaneous Functions
Sequence Manipulation
Several real-time applications running on the user's workstation accept, manipulate, and return nucleic acid sequence strings (strings of characters). Functions available in this group perform sequence creation, manipulation via several types of mutations (single or many bases), and translation to amino acids sequences . A typical sequence format, among several possible, is illustrated below:
;test.seq file - sequence in STANFORD format
m2
GAAUUACCGAUAUCGAUACAUCAGGAAUAUUUGAUUCAGAUGAUAUGACUAUCAAGGCCG
CCUGAGUGCGGUUUUACCGCAUACCAAUAACGCUUCACUCGAGGCGUUUUUCGUUAUGUA
UAAAUAAGAAGCACACCAUGCAAUAUGCCAUUGCAGGGUGGCCUGUUGCUGGCUGCCCUU
CCGAAUCUUUACUUGAACGAA1
A CT file format (MFOLD 3.0+ output format) that contains both sequence and structure information, is illustrated below:
First line: sequence fragment length (L), free energy, sequence name, Subsequent lines (n-th records): n (1 thru L), n-th nucleotide, 5'-connecting base index (n-1), 3'-connecting base index (n+1), paired base index, and the n-th base index in the original sequence, i.e. absolute seq. positions (fold start, start+L)
If the value of 5', 3' or paired base index is zero, it means that the nucleotide is not connected or paired with anything.
314 dG = -98.3 rabbit-RBG-mRNA
| 1 | G | 0 | 2 | 47 | 10 |
| 2 | G | 1 | 3 | 46 | 11 |
| 3 | G | 2 | 4 | 45 | 12 |
| 4 | A | 3 | 5 | 0 | 13 |
| 5 | G | 4 | 6 | 43 | 14 |
| : | : | : | : | : | : |
Folding Algorithms
Folding programs employ two different types of algorithms; the Dynamic Programming Algorithm (DPA) (references)and the Genetic Algorithm (GA)(references). While both attempt to predict secondary structures of an RNA sequence, they differ in basic concepts used. The RNA folding algorithms accept sequence files (strings) as input, and output multiple region tables indicating which bases (nucleotides) are paired in a folded structure. These region tables reflect energetically optimal and suboptimal solutions based on standardized energy rules.
Region table representation:
| Region number |
Start (First 5' base) |
Stop (Last 3' base) |
Region size (# of base pairs) | Energy (kcal/mol) |
| 1) | 9 | 53 | 4 | -4.3 |
| 2) | 13 | 48 | 2 | -2.3 |
| 3) | 16 | 46 | 3 | -2.0 |
| : | : | : | : | : |
Structure Representations
Structural representation for a set of RNA molecules can be created based on the region files created by folding programs. Secondary (2D) structures can also be represented as trees, utilizing Lisp's nested list notation, with symbols such as M (multibranch loop), B (bulge loop), I (internal loop), and H (hairpin loop). Optional R's present in some representations are no-op place holders indicating regions (hence R) or stems. The tree representation facilitates multiple levels of abstraction of the actual structure allowing for structural comparisons of varying strictness by taxonomy tree clustering, based upon measures of structure similarity. In addition, multiple alignment methods and structural motifs matching can be employed.
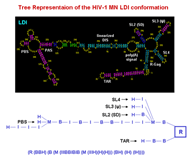
Two of the Tree List representations used are shown below:
(N(H)(H)(BH)(H)(H)(H)(BBBIH)) - condensed
(N(R(H))(R(H))(R(B(R(H))))(R(H))(R(H))(R(H)) - expanded (explicit)
(R(B(R(B(R(B(R(I(R(H)))))))))))
Structure Alignment
A pair-wise 'Needleman-Wunsch' alignment function permits clustering of RNA secondary structures based upon similarity of substructures. It uses the parenthesized string form for representing trees, described earlier, and performs a multiple alignment clustering of such representations (Shapiro 1988).
Taxonomy Operations
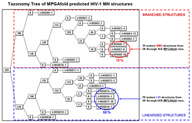
Taxonomy tree applications form a class of functions permitting calculation, display, search, and manipulation of structural taxonomy trees. One application does a pair-wise tree comparison on the tree representations (mentioned above) of the RNA secondary structures and generates a taxonomy tree which clusters the structures based upon heuristic measures of similarity (Shapiro and Zhang 1990). Another function draws the tree and facilitates its manipulation (Shapiro and Kasprzak 1996).
Structure Matching
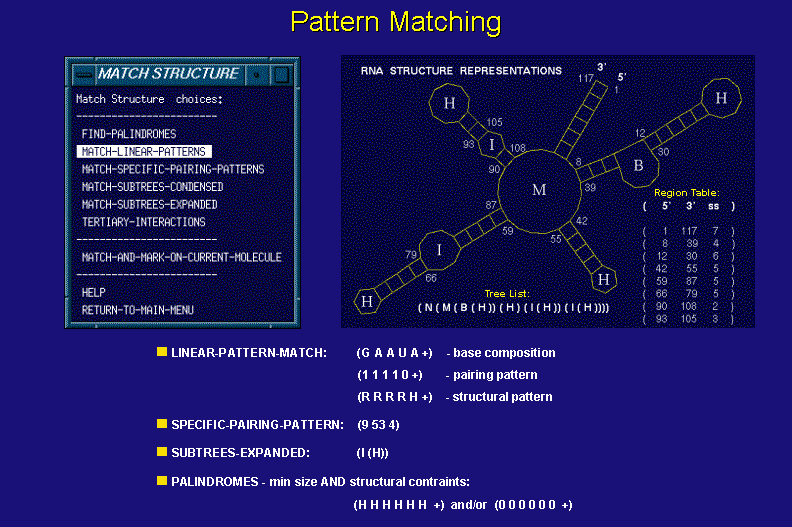
The structure matching class of functions deals with motif analysis of a set of structures (possibly thousands). Functions available in this class can be divided into two subclasses dealing with structure matching and linear features matching. The structure pattern matching operates on the tree list representations and performs the pattern searches for structural motif queries which may include wildcards.
{kind=link}
The linear pattern matching functions utilize data structures used in RNA structure drawings. They allow one to perform base-by base searches on conjunctions of linear patterns including sequence, pairing, and structural element membership of the input sequence and its structural conformations.
RNA Structure Drawing and Manipulation (2D and 3D)
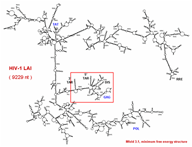
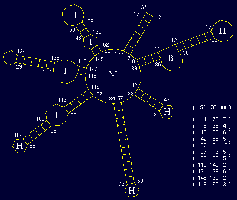
This functional domain supports drawing of RNA secondary structures and manipulation of the actual drawings for optimum visualization. (rotation, resizing, bending/untangling, labeling, annotating) The drawings are based on a sequence file and related region tables. In addition, drawings can be generated directly from stem histograms (composite drawings) or stem traces, both described below. There is also a three-dimensional visualization and analysis tool utilizing H. Martinez's rna_2d3d software, which generates 3D atomic coordinates from structures predicted by the various folding programs (H. Martinez, personal communication). RasMol, and Midas are used for visualization of these three-dimensional representations.
The two small figures to the right are of full genome fold of HIV-1 RF. The figure on the far right shows the structure drawing after it has been untangled; first via an automatic untangling tool, then custom shaped via an interactive untangler.
Two-Dimensional Stem Histogram (Dot Matrix)
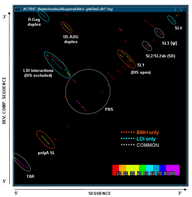
This function produces a two-dimensional histogram of all base pairs that exist in a set of suboptimal structures produced by one of the RNA folding programs. In other words, a two-dimensional histogram gives a good picture of how prevalent certain structural motifs are among large numbers of energetically optimal and suboptimal structures. Combining the statistical base pairing matrix analysis with the energy-oriented significance/stability information and stem trace (see below) helps to get a fuller picture of the reliability of molecule folding predictions.
Stem Trace
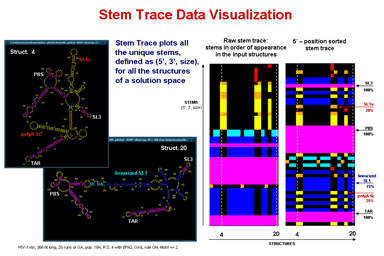
This function produces a two-dimensional plot of all unique regions (stems), defined as triplets (5'-position, 3'-position, stem-size), that exist in a set of structures (regions files). It is orthogonal to a stem histogram of the same data. Whereas a stem histogram stresses the cumulative nature of results and loses associations between particular stems and structures from the plotted solution space, a stem trace of the same data explicitly depicts all the individual structure-stem relationships. What is lost visually in the Stem Trace plots are spatial relations between stems that are immediately obvious in a stem histogram. However, This data (5', 3',and size values), together with other information, is automaticaly displayed in the Stem Trace Control Window as the user moves a mouse pointer over the plot.
Stem Trace can be used in the analysis of GA (Genetic Algorithm) structure predictions and for visual exploration of the space of suboptimal solutions predicted by the DPA (Dynamic Programming Algorithm) folding programs (Kasprzak and Shapiro 1999, Shapiro et al. 2001b, Atha et al. 2001, Kasprzak and Shapiro 2002). The horizontal axis shows generations (for GA traces) or suboptimal solutions (in case of DPA), and the vertical axis shows unique regions. Persistence of structural elements thus can be viewed.
Stem trace can be used to analyze the following major types of inputs:
- data from each generation of one run of the GA for RNA folding;
- sampled as a structure from a specified processor
- sampled as a peak histogram representative
- the best results for multiple GA runs for one input sequence;
- may be grouped by folding conditions, such as population size
- ordered sequence of suboptimal results generated by the DPA, such as MFOLD;
- any of the above for multiple sequences of a family.
Tertiary Interactions Prediction
This utility generates lists of all potential tertiary interactions between the elements of the predicted two-dimensional (secondary) RNA structure (i.e. for a specified sequence and a related region table). Included in these are some valid pseudoknots. The predicted interactions may be filtered based on the user selected criteria.
Miscellaneous Functions
A variety of miscellaneous functions is provided to let the user perform "housekeeping" and monitoring tasks not necessarily strictly related to any specific functional domain but available in many of them.
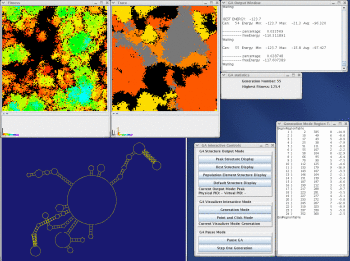
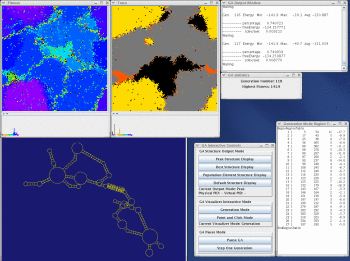
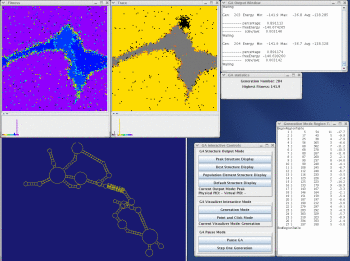
Take a look at the MPGAfold, MPGAfold Visualizer, and StructureLab demo displaying an RNA folding pathway.
For information on obtaining a copy of STRUCTURELAB, please click here.